About Fabry disease
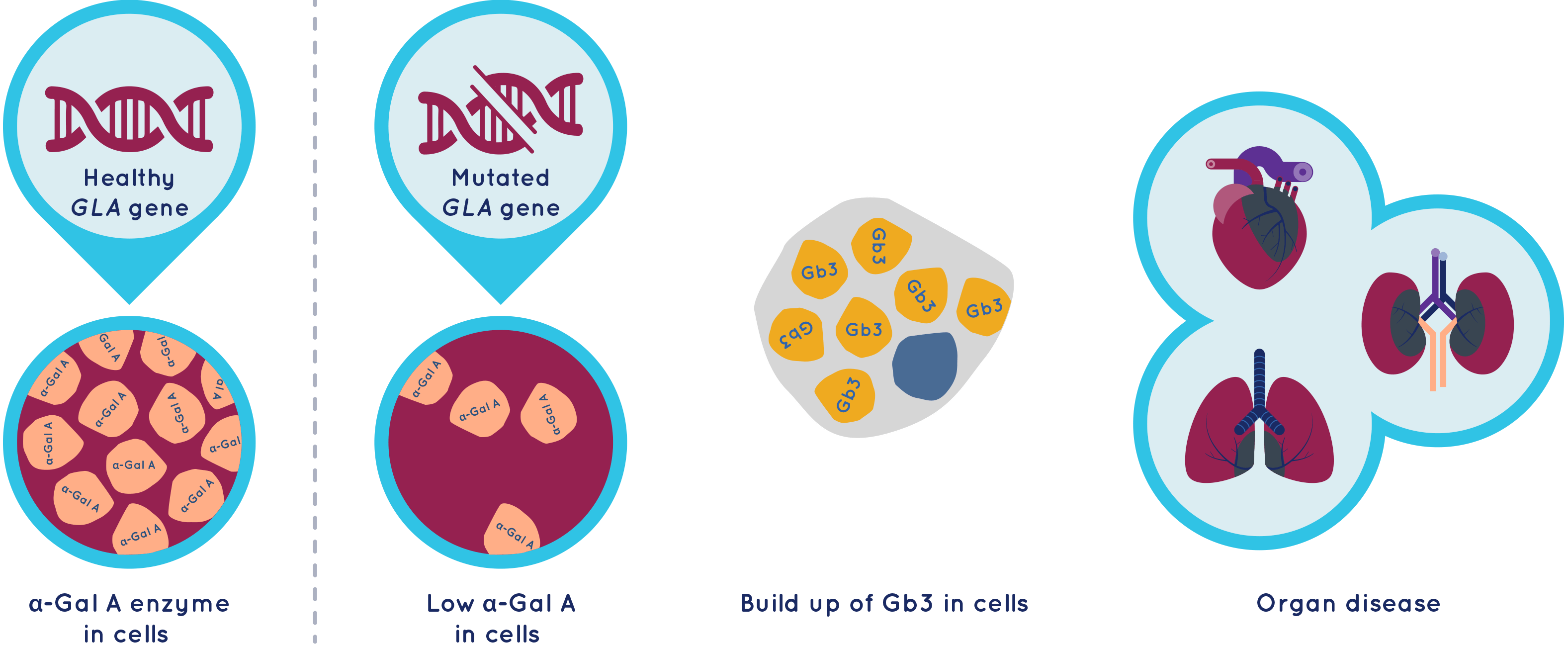
Fabry disease is caused by changes (mutations) in the galactosidase alpha (GLA) gene, which is responsible for providing the body with instructions for producing alpha-galactosidase A (α-Gal A) enzyme. The α-Gal A enzyme is needed to process and get rid of a fatty substance called globotriaosylceramide (Gb3) from cells. If the body can’t get rid of Gb3 then it can lead to serious disease of vital organs.
Signs and symptoms of Fabry disease
There are two types of Fabry disease: classical and non-classical disease, which can occur in both males and females.
People with classical Fabry disease produce little or no α-Gal A enzyme.
Symptoms are usually first experienced in early childhood. The average age of the first symptoms in males and females is 10.5 years, and yet the average age of diagnosis is 28.5 years. Diagnosing the disease can be difficult because of the range of symptoms that can occur over time. Early symptoms can include:
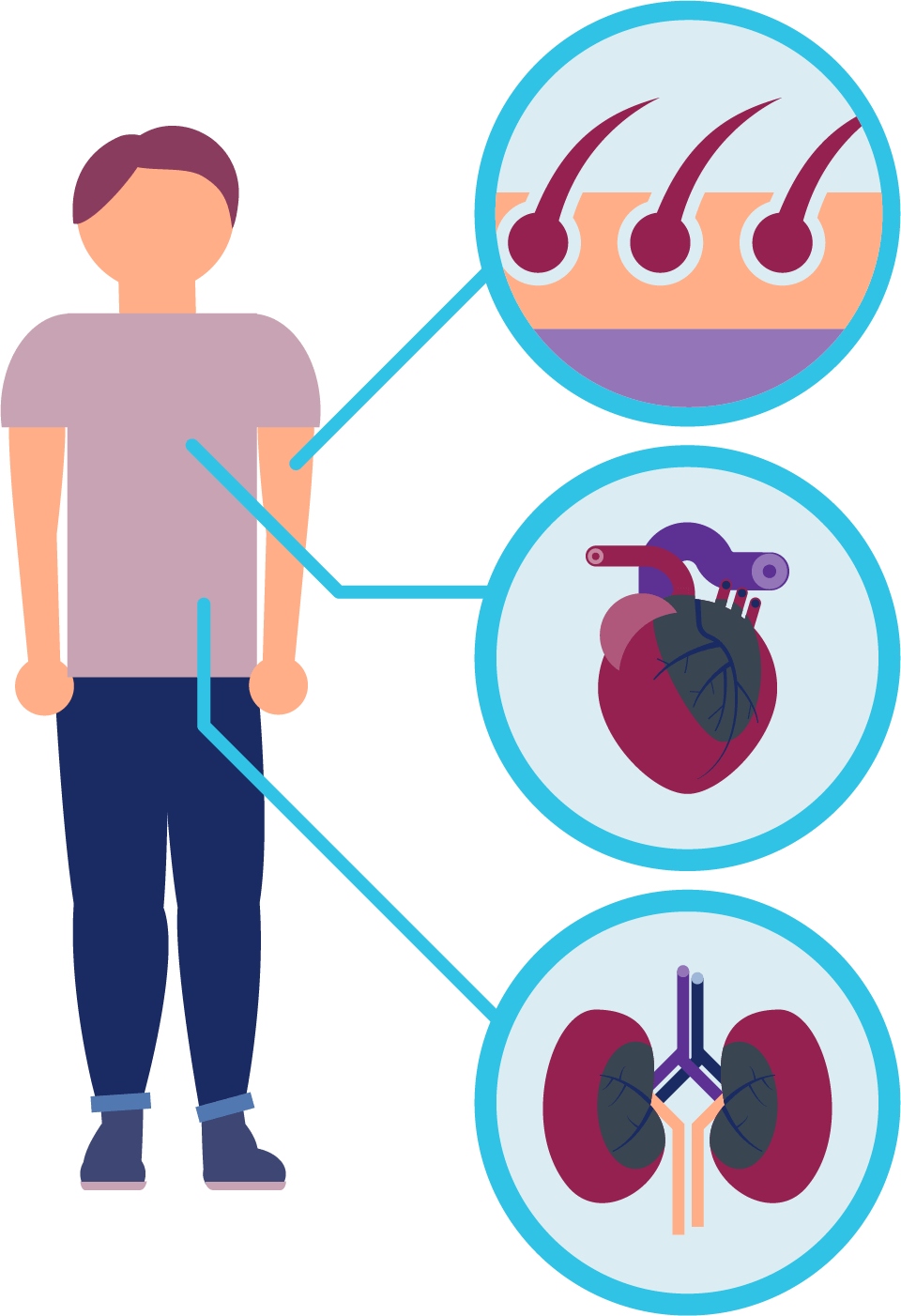
- Episodes of pain and burning sensations [known medically as neuropathic pain]
- Decreased or absent sweat production and discomfort (heat intolerance) in warm temperatures, with exercise or fevers [anhidrosis or hypohidrosis]
- Development of small, raised reddish purple blemishes on the skin [angiokeratoma]
- Cloudiness of the front surface (cornea) of the eye [cornea verticillata]
The disease gets progressively worse over time and can affect many vital organs, including the kidneys, heart, nerves, skin, eyes and gut.
People with non-classical Fabry disease, also called late-onset or atypical Fabry disease, have low levels of α-Gal A enzyme. They are generally less severely affected and the disease may be limited to a single organ.
For more information on Fabry disease please visit:



Treatment
Current treatments for Fabry disease aim to increase the level of active α-Gal A in the bloodstream.
Enzyme replacement therapy (ERT)
- Uses ‘man-made’ versions of α-Gal A
- Its goal is to replace the missing α-Gal A enzyme in the blood
- This then lowers the amount of a fatty substance called globotriaosylceramide (Gb3) that build-up in tissues
- Current ERT involves regular infusions via a vein with a needle or catheter, usually once every two weeks
Chaperone therapy
- Works in 40–50% of patients with certain genetic faults found on the α-Gal A enzyme
- It stabilizes the α-Gal A enzyme so it can then do its job of clearing Gb3 from the blood and reduce the build-up in tissue
Scientific research
Several research studies are currently underway to explore other potential new treatment approaches, including investigational gene therapy.

Other treatments for Fabry disease are aimed at relieving specific symptoms
Treatments that can relieve the symptoms of Fabry disease include painkillers, anti-clotting drugs to prevent strokes and drugs to treat heart disease.
In severe Fabry disease that has progressed to kidney failure, hemodialysis and kidney transplantation may be necessary.